




Abstract
Machine learning is a useful computational tool for large and complex tasks such as those in the field of enzyme engineering, selection and design. In this review, we examine enzyme-related applications of machine learning. We start by comparing tools that can identify the function of an enzyme and the site responsible for that function. Then we detail methods for optimizing important experimental properties, such as the enzyme environment and enzyme reactants. We describe recent advances in enzyme systems design and enzyme design itself. Throughout we compare and contrast the data and algorithms used for these tasks to illustrate how the algorithms and data can be best used by future designers.
Introduction
Enzymes catalyze chemical reactions that would otherwise require high temperature or pressure, increasing their reaction rates up to a million-fold. Because of these qualities, enzymes find numerous applications in environmental remediation, (Sharma et al., 2018) human health and industrial synthesis (Jegannathan and Nielsen, 2013). Moreover, using enzymes in industrial synthesis is often more environmentally friendly than other synthetic alternatives (Jegannathan and Nielsen, 2013). Designing, engineering and selecting enzymes is therefore a subject of great medical, environmental and industrial importance.
A large number of enzymes have already found their way into scientific research and industry. With a sense that the properties of these natural proteins could be improved, decades-long design efforts have continually modified these enzymes in order to sharpen their function and optimize the conditions under which they work (Fox and Huisman, 2008). Overall, protein design has promised not just new proteins but also that the process of designing new proteins would reveal fundamental truths about protein folding and protein interactions. The same is true for enzyme design as it can teach what intrinsic enzymatic properties can be found in the sequences of amino acids and in the atomic arrangements of catalytic sites. However, sequence space is very large, and nature has left a significant part of it unexplored. It is likely that new useful enzymes could be created if we could explore these untouched regions of the protein sequence space in an efficient manner.
Machine learning (ML) is becoming the tool of choice for such exploratory efforts. ML is learning done without full, manual oversight. ML is also sometimes called statistical learning because it focuses on patterns or statistical properties of data. ML can mean learning the distribution of the data, learning to make predictions for new data, learning to generate new data from estimated data distributions, or even the process of continuous learning by machines, as in artificial intelligence. As it applies to enzyme engineering, ML provides a way to use biological data (at the level of organism, protein sequence, protein structure, residue or atoms) to extract information (patterns or data distributions) that can then be used for downstream tasks such as classifying new enzymes, predicting properties for enzymes or their substrates, predicting the optimal microenvironment and finding new enzymes or combination of enzymes that have better catalytic activity. In protein engineering, ML can help model the important sequence-to-function and structure-to-function relationships that we still need to understand more fully (Siedhoff et al., 2020).
The ML umbrella also encompasses another term, deep learning. Deep models are networks of large size and complexity that are able to model difficult data distributions such as data of high dimensionality. These models extract useful patterns directly from the raw data without the need for manually created features or any human intervention. However, due to the large number of parameters in deep models, much more training data are required for model learning. With more experimental data and more computational power available, ML efforts in the field of enzymology will grow, including the creation of new databases for training, data preprocessing (Mazurenko et al., 2020) and the ethical concerns related to creating new biomolecules with unintended characteristics (Kemp et al., 2020).
Over the past three years ML has been applied toward answering long standing questions in enzymology: predicting the function (or functions) of a putative enzyme given its sequence or structure, predicting which mutations would improve catalytic turnover rates and predicting how environmental changes will affect enzyme function. Here we review those recent advances specifically focusing on the preferred datasets, algorithms and features that facilitated the advances of the past three years in enzyme identification, classification, optimization, systemization and finally, de novo enzyme design. Special consideration is given to models that are interpretable such that the importance of features to enzyme function is revealed. The top models we discuss are summarized in Table I.
Enzyme Function Prediction
A substantial amount of work has been done in attempting to computationally predict enzymatic classification and catalytic site location. Common non-ML methodology for both includes using homology to detect enzyme function from the entire sequence (BLAST+) (Camacho et al., 2009), sequence motif (PROSITE) (Sigrist et al., 2013) or a domain (Pfam) (Finn et al., 2016). These tools predict enzyme function by using previously annotated enzymes or enzyme sites and find sequences or sites that are similar to the annotated example. The homology-based predictions are not sensitive to small, important amino acid changes and require numerous, well-studied homologs. In contrast, ML methods use sequence and structural features outside of sequence or structural similarity, allowing ML models to make accurate enzymatic predictions on proteins with few or no homologs.
ML for enzymatic classification
The Nomenclature Committee of the International Union of Biochemistry classifies enzymatic reactions using a four-level hierarchy, called Enzyme Commission (EC) numbers (McDonald and Tipton, 2014). EC numbers contain four numbers, one for each level, separated by periods (e.g. 1.2.3.4). The task of predicting if a protein is enzymatic or not enzymatic is often referred to as level zero. Level one divides enzymes into seven major enzyme classes; 1: oxidoreductases, 2: transferases, 3: hydrolases, 4: lyases, 5: isomerases, 6: ligases and 7: translocases. The next three levels use reaction elements such as the chemical bond and substrate to further categorize enzymes into different subclasses. Previous ML EC number predictors have used classical classification algorithms (Shen and Chou, 2007; De Ferrari et al., 2012; Kumar and Skolnick, 2012; Che et al., 2016; Li et al., 2016; Zou and Xiao, 2016; Amidi et al., 2017). These classical algorithms differ from the current mostly deep learning predictors in that the classical predictors require user defined features. Conversely, deep learning methods extract features from raw data representations, which lead to overall better metrics. However, deep models require substantially more data and can therefore underperform when data is scant. Moreover, due to the opacity of deep learning models further troubleshooting of poor performing models is challenging (Waldrop, 2019). The transition to and popularity of deep networks coincided with availability of deep learning methods in TensorFlow (Martín et al., 2015). Here we compare the similarities and differences of four recent and effective ML EC number predictors: DEEPre (Li et al., 2018), mlDEEPre (Zou et al., 2019), ECPred (Dalkiran et al., 2018) and DeepEC (Ryu et al., 2019).
Resource table. Selected list of recent, excellent enzymatic ML tools and methods organized by the task they perform and the corresponding section of the review in which they are covered (some tools have additional tasks). A dash is used in lieu of a name if a tool/method that does not have an author designated name. ML method abbreviations are: convolutional neural network (CNN), support vector machine (SVM), k-nearest neighbors (kNN), recurrent neural networks (RNN), random forest (RF), naïve Bayes (NB), gradient boosting regression trees (GB), partial least squares regression (PLSR), linear regression (LR), multi-layer perceptron (MLP), neural network (NN) and direct coupling analysis (DCA). Input type abbreviations are: sequence (Seq), structure (Struct) and experimental (Exp). Best use specifies what each resource is capable of predicting, highlighting differences between similar resources when applicable. Availability specifies if a resource tool could be accessed via a webserver or if the model/code is available for download. All the tools with available webserver also have download options
| Resource | Task | ML method | Input type | Best use | Availability | Citation |
|---|---|---|---|---|---|---|
| DeepEC | Enzymatic Classification | CNN | Seq | Complete EC number prediction | Downloadable | Ryu et al., 2019 |
| ECPred | Enzymatic classification | Ensemble (SVMs, kNN) | Seq | Complete or partial EC number prediction | Webserver | Dalkiran et al., 2018 |
| mlDEEPre | Enzymatic classification | Ensemble (CNN, RNN) | Seq | Multiple EC number predictions for one sequence | Webserver | Zou et al., 2019 |
| PREvaIL | Enzyme site prediction | RF | Seq, Struct | Predicting catalytic residues in a protein structure | Downloadable | Song et al., 2018 |
| 3DCNN | Enzyme site prediction | CNN | Struct | Describes method for predicting specific enzyme reaction site | No | Torng and Altman, 2019 |
| MAHOMES | Enzyme site prediction | RF | Struct | Predicting catalytic metal ions bound to protein | Downloadable | Feehan et al., 2021 |
| POOL | Enzyme site prediction | POOL | Seq, Struct | Predicting catalytic residues in a protein structure | Webserver | Somarowthu et al., 2011 |
| TOME | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures | Downloadable | Li et al., 2019a, b |
| TOMER | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures for imbalanced data | Downloadable | Gado et al., 2020 |
| TAXyl | Condition optimization | RF | Seq | Classify thermophilicity of an enzyme | Downloadable | Foroozandeh Shahraki et al., 2020 |
| SoluProt | Condition optimization | RF | Seq | Predict enzyme solubility in E. coli expression system | Webserver | Hon et al., 2020 |
| POOL | Substrate identification | NB | Seq | Predict/design transferase substrates with orthogonal binding | Downloadable | Tallorin et al., 2018 |
| – | Substrate identification | GB | Seq | Predict glycosyltransferase sugar specificity | No | Taujale et al., 2020 |
| pNPred | Substrate identification | RF | Seq, Struct | Substrate specificity of thiolases towards p-nitrophenyl esters | Webserver | Robinson, Smith et al., 2020a |
| AdenylPred | Substrate identification | RF | Seq, Struct | Functional and substrate classification of adenylate-forming enzymes | Webserver | Robinson, Terlouw et al., 2020b |
| innov’SAR | Substrate identification | PLSR | Seq | Reduce the size of mutant libraries required for successful directed evolution of epoxide hydrolase enzyme stereoselectivity | No | Cadet et al., 2018 |
| innov’SAR | Turnover rate | PLSR | Seq | Optimal combination of mutations to improve activity | No | Ostafe et al., 2020 |
| innov’SAR | Stability | PLSR | Seq | Identify combinations of limonene epoxide hydrolase mutations that are less prone to aggregation | No | Li et al., 2021 |
| – | Turnover rate | LR | Struct | Determine reactivity of substrate-enzyme pair | No | Bonk et al., 2019 |
| SolventNet | Turnover rate | CNN | Struct | Hydrolysis rate effects of acid catalyst and solvents | No | Chew et al., 2020 |
| ART/EVOLVE | Systems engineering | Ensemble (Bayesian) | Exp | Predict combination of promoters that improve Trp metabolism | Downloadable | Zhang et al., 2020 |
| GC-ANN | Systems engineering | MLP | Exp | Pathway flux from concentrations of upstream enzymes | Downloadable | Ajjolli Nagaraja et al., 2020 |
| iPROBE | Systems engineering | Ensemble (MLP) | Seq | Pathway yield from concentrations of upstream enzymes | No | Karim et al., 2020 |
| – | Systems engineering | Ensemble (RF, NN, ElasticNet) | Exp, Struct | Predict enzyme turnover numbers for genome-scale models | Upon request | Heckmann et al., 2018 |
| – | Systems engineering | MDP, NN | Exp | Predict regulation level of enzymes in metabolic models | No | Britton et al., 2020 |
| bmDCA | Design | DCA | Seq | Design new sequences with chorismate mutase function | Downloadable | Russ et al., 2020 |
| – | Design | LR | Struct | Design aldehyde deformylating oxygenase function on non-functional alpha-bundle scaffolds | Downloadable | Mak et al., 2020 |
| Resource | Task | ML method | Input type | Best use | Availability | Citation |
|---|---|---|---|---|---|---|
| DeepEC | Enzymatic Classification | CNN | Seq | Complete EC number prediction | Downloadable | Ryu et al., 2019 |
| ECPred | Enzymatic classification | Ensemble (SVMs, kNN) | Seq | Complete or partial EC number prediction | Webserver | Dalkiran et al., 2018 |
| mlDEEPre | Enzymatic classification | Ensemble (CNN, RNN) | Seq | Multiple EC number predictions for one sequence | Webserver | Zou et al., 2019 |
| PREvaIL | Enzyme site prediction | RF | Seq, Struct | Predicting catalytic residues in a protein structure | Downloadable | Song et al., 2018 |
| 3DCNN | Enzyme site prediction | CNN | Struct | Describes method for predicting specific enzyme reaction site | No | Torng and Altman, 2019 |
| MAHOMES | Enzyme site prediction | RF | Struct | Predicting catalytic metal ions bound to protein | Downloadable | Feehan et al., 2021 |
| POOL | Enzyme site prediction | POOL | Seq, Struct | Predicting catalytic residues in a protein structure | Webserver | Somarowthu et al., 2011 |
| TOME | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures | Downloadable | Li et al., 2019a, b |
| TOMER | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures for imbalanced data | Downloadable | Gado et al., 2020 |
| TAXyl | Condition optimization | RF | Seq | Classify thermophilicity of an enzyme | Downloadable | Foroozandeh Shahraki et al., 2020 |
| SoluProt | Condition optimization | RF | Seq | Predict enzyme solubility in E. coli expression system | Webserver | Hon et al., 2020 |
| POOL | Substrate identification | NB | Seq | Predict/design transferase substrates with orthogonal binding | Downloadable | Tallorin et al., 2018 |
| – | Substrate identification | GB | Seq | Predict glycosyltransferase sugar specificity | No | Taujale et al., 2020 |
| pNPred | Substrate identification | RF | Seq, Struct | Substrate specificity of thiolases towards p-nitrophenyl esters | Webserver | Robinson, Smith et al., 2020a |
| AdenylPred | Substrate identification | RF | Seq, Struct | Functional and substrate classification of adenylate-forming enzymes | Webserver | Robinson, Terlouw et al., 2020b |
| innov’SAR | Substrate identification | PLSR | Seq | Reduce the size of mutant libraries required for successful directed evolution of epoxide hydrolase enzyme stereoselectivity | No | Cadet et al., 2018 |
| innov’SAR | Turnover rate | PLSR | Seq | Optimal combination of mutations to improve activity | No | Ostafe et al., 2020 |
| innov’SAR | Stability | PLSR | Seq | Identify combinations of limonene epoxide hydrolase mutations that are less prone to aggregation | No | Li et al., 2021 |
| – | Turnover rate | LR | Struct | Determine reactivity of substrate-enzyme pair | No | Bonk et al., 2019 |
| SolventNet | Turnover rate | CNN | Struct | Hydrolysis rate effects of acid catalyst and solvents | No | Chew et al., 2020 |
| ART/EVOLVE | Systems engineering | Ensemble (Bayesian) | Exp | Predict combination of promoters that improve Trp metabolism | Downloadable | Zhang et al., 2020 |
| GC-ANN | Systems engineering | MLP | Exp | Pathway flux from concentrations of upstream enzymes | Downloadable | Ajjolli Nagaraja et al., 2020 |
| iPROBE | Systems engineering | Ensemble (MLP) | Seq | Pathway yield from concentrations of upstream enzymes | No | Karim et al., 2020 |
| – | Systems engineering | Ensemble (RF, NN, ElasticNet) | Exp, Struct | Predict enzyme turnover numbers for genome-scale models | Upon request | Heckmann et al., 2018 |
| – | Systems engineering | MDP, NN | Exp | Predict regulation level of enzymes in metabolic models | No | Britton et al., 2020 |
| bmDCA | Design | DCA | Seq | Design new sequences with chorismate mutase function | Downloadable | Russ et al., 2020 |
| – | Design | LR | Struct | Design aldehyde deformylating oxygenase function on non-functional alpha-bundle scaffolds | Downloadable | Mak et al., 2020 |
Resource table. Selected list of recent, excellent enzymatic ML tools and methods organized by the task they perform and the corresponding section of the review in which they are covered (some tools have additional tasks). A dash is used in lieu of a name if a tool/method that does not have an author designated name. ML method abbreviations are: convolutional neural network (CNN), support vector machine (SVM), k-nearest neighbors (kNN), recurrent neural networks (RNN), random forest (RF), naïve Bayes (NB), gradient boosting regression trees (GB), partial least squares regression (PLSR), linear regression (LR), multi-layer perceptron (MLP), neural network (NN) and direct coupling analysis (DCA). Input type abbreviations are: sequence (Seq), structure (Struct) and experimental (Exp). Best use specifies what each resource is capable of predicting, highlighting differences between similar resources when applicable. Availability specifies if a resource tool could be accessed via a webserver or if the model/code is available for download. All the tools with available webserver also have download options
| Resource | Task | ML method | Input type | Best use | Availability | Citation |
|---|---|---|---|---|---|---|
| DeepEC | Enzymatic Classification | CNN | Seq | Complete EC number prediction | Downloadable | Ryu et al., 2019 |
| ECPred | Enzymatic classification | Ensemble (SVMs, kNN) | Seq | Complete or partial EC number prediction | Webserver | Dalkiran et al., 2018 |
| mlDEEPre | Enzymatic classification | Ensemble (CNN, RNN) | Seq | Multiple EC number predictions for one sequence | Webserver | Zou et al., 2019 |
| PREvaIL | Enzyme site prediction | RF | Seq, Struct | Predicting catalytic residues in a protein structure | Downloadable | Song et al., 2018 |
| 3DCNN | Enzyme site prediction | CNN | Struct | Describes method for predicting specific enzyme reaction site | No | Torng and Altman, 2019 |
| MAHOMES | Enzyme site prediction | RF | Struct | Predicting catalytic metal ions bound to protein | Downloadable | Feehan et al., 2021 |
| POOL | Enzyme site prediction | POOL | Seq, Struct | Predicting catalytic residues in a protein structure | Webserver | Somarowthu et al., 2011 |
| TOME | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures | Downloadable | Li et al., 2019a, b |
| TOMER | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures for imbalanced data | Downloadable | Gado et al., 2020 |
| TAXyl | Condition optimization | RF | Seq | Classify thermophilicity of an enzyme | Downloadable | Foroozandeh Shahraki et al., 2020 |
| SoluProt | Condition optimization | RF | Seq | Predict enzyme solubility in E. coli expression system | Webserver | Hon et al., 2020 |
| POOL | Substrate identification | NB | Seq | Predict/design transferase substrates with orthogonal binding | Downloadable | Tallorin et al., 2018 |
| – | Substrate identification | GB | Seq | Predict glycosyltransferase sugar specificity | No | Taujale et al., 2020 |
| pNPred | Substrate identification | RF | Seq, Struct | Substrate specificity of thiolases towards p-nitrophenyl esters | Webserver | Robinson, Smith et al., 2020a |
| AdenylPred | Substrate identification | RF | Seq, Struct | Functional and substrate classification of adenylate-forming enzymes | Webserver | Robinson, Terlouw et al., 2020b |
| innov’SAR | Substrate identification | PLSR | Seq | Reduce the size of mutant libraries required for successful directed evolution of epoxide hydrolase enzyme stereoselectivity | No | Cadet et al., 2018 |
| innov’SAR | Turnover rate | PLSR | Seq | Optimal combination of mutations to improve activity | No | Ostafe et al., 2020 |
| innov’SAR | Stability | PLSR | Seq | Identify combinations of limonene epoxide hydrolase mutations that are less prone to aggregation | No | Li et al., 2021 |
| – | Turnover rate | LR | Struct | Determine reactivity of substrate-enzyme pair | No | Bonk et al., 2019 |
| SolventNet | Turnover rate | CNN | Struct | Hydrolysis rate effects of acid catalyst and solvents | No | Chew et al., 2020 |
| ART/EVOLVE | Systems engineering | Ensemble (Bayesian) | Exp | Predict combination of promoters that improve Trp metabolism | Downloadable | Zhang et al., 2020 |
| GC-ANN | Systems engineering | MLP | Exp | Pathway flux from concentrations of upstream enzymes | Downloadable | Ajjolli Nagaraja et al., 2020 |
| iPROBE | Systems engineering | Ensemble (MLP) | Seq | Pathway yield from concentrations of upstream enzymes | No | Karim et al., 2020 |
| – | Systems engineering | Ensemble (RF, NN, ElasticNet) | Exp, Struct | Predict enzyme turnover numbers for genome-scale models | Upon request | Heckmann et al., 2018 |
| – | Systems engineering | MDP, NN | Exp | Predict regulation level of enzymes in metabolic models | No | Britton et al., 2020 |
| bmDCA | Design | DCA | Seq | Design new sequences with chorismate mutase function | Downloadable | Russ et al., 2020 |
| – | Design | LR | Struct | Design aldehyde deformylating oxygenase function on non-functional alpha-bundle scaffolds | Downloadable | Mak et al., 2020 |
| Resource | Task | ML method | Input type | Best use | Availability | Citation |
|---|---|---|---|---|---|---|
| DeepEC | Enzymatic Classification | CNN | Seq | Complete EC number prediction | Downloadable | Ryu et al., 2019 |
| ECPred | Enzymatic classification | Ensemble (SVMs, kNN) | Seq | Complete or partial EC number prediction | Webserver | Dalkiran et al., 2018 |
| mlDEEPre | Enzymatic classification | Ensemble (CNN, RNN) | Seq | Multiple EC number predictions for one sequence | Webserver | Zou et al., 2019 |
| PREvaIL | Enzyme site prediction | RF | Seq, Struct | Predicting catalytic residues in a protein structure | Downloadable | Song et al., 2018 |
| 3DCNN | Enzyme site prediction | CNN | Struct | Describes method for predicting specific enzyme reaction site | No | Torng and Altman, 2019 |
| MAHOMES | Enzyme site prediction | RF | Struct | Predicting catalytic metal ions bound to protein | Downloadable | Feehan et al., 2021 |
| POOL | Enzyme site prediction | POOL | Seq, Struct | Predicting catalytic residues in a protein structure | Webserver | Somarowthu et al., 2011 |
| TOME | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures | Downloadable | Li et al., 2019a, b |
| TOMER | Condition optimization | RF | Seq | Predict optimal growth and catalytic temperatures for imbalanced data | Downloadable | Gado et al., 2020 |
| TAXyl | Condition optimization | RF | Seq | Classify thermophilicity of an enzyme | Downloadable | Foroozandeh Shahraki et al., 2020 |
| SoluProt | Condition optimization | RF | Seq | Predict enzyme solubility in E. coli expression system | Webserver | Hon et al., 2020 |
| POOL | Substrate identification | NB | Seq | Predict/design transferase substrates with orthogonal binding | Downloadable | Tallorin et al., 2018 |
| – | Substrate identification | GB | Seq | Predict glycosyltransferase sugar specificity | No | Taujale et al., 2020 |
| pNPred | Substrate identification | RF | Seq, Struct | Substrate specificity of thiolases towards p-nitrophenyl esters | Webserver | Robinson, Smith et al., 2020a |
| AdenylPred | Substrate identification | RF | Seq, Struct | Functional and substrate classification of adenylate-forming enzymes | Webserver | Robinson, Terlouw et al., 2020b |
| innov’SAR | Substrate identification | PLSR | Seq | Reduce the size of mutant libraries required for successful directed evolution of epoxide hydrolase enzyme stereoselectivity | No | Cadet et al., 2018 |
| innov’SAR | Turnover rate | PLSR | Seq | Optimal combination of mutations to improve activity | No | Ostafe et al., 2020 |
| innov’SAR | Stability | PLSR | Seq | Identify combinations of limonene epoxide hydrolase mutations that are less prone to aggregation | No | Li et al., 2021 |
| – | Turnover rate | LR | Struct | Determine reactivity of substrate-enzyme pair | No | Bonk et al., 2019 |
| SolventNet | Turnover rate | CNN | Struct | Hydrolysis rate effects of acid catalyst and solvents | No | Chew et al., 2020 |
| ART/EVOLVE | Systems engineering | Ensemble (Bayesian) | Exp | Predict combination of promoters that improve Trp metabolism | Downloadable | Zhang et al., 2020 |
| GC-ANN | Systems engineering | MLP | Exp | Pathway flux from concentrations of upstream enzymes | Downloadable | Ajjolli Nagaraja et al., 2020 |
| iPROBE | Systems engineering | Ensemble (MLP) | Seq | Pathway yield from concentrations of upstream enzymes | No | Karim et al., 2020 |
| – | Systems engineering | Ensemble (RF, NN, ElasticNet) | Exp, Struct | Predict enzyme turnover numbers for genome-scale models | Upon request | Heckmann et al., 2018 |
| – | Systems engineering | MDP, NN | Exp | Predict regulation level of enzymes in metabolic models | No | Britton et al., 2020 |
| bmDCA | Design | DCA | Seq | Design new sequences with chorismate mutase function | Downloadable | Russ et al., 2020 |
| – | Design | LR | Struct | Design aldehyde deformylating oxygenase function on non-functional alpha-bundle scaffolds | Downloadable | Mak et al., 2020 |
ML methods for classification of enzyme reactions are mostly sequence-based, due in large part to the abundance of protein sequences with associated EC numbers. Some of these models use homologous sequences in order to have more data while other models avoid homologs to prevent overfitting. For example, DeepEC uses 1 388 606 protein sequences with 4669 different EC numbers. The data set was made by mapping EC numbers to over a million unannotated sequences using sequence similarity. Although homologs were seen as helpful for training DeepEC, it is more common to remove sequences with high similarity in order to avoid bias and overfitting during performance evaluation. For example, another ML enzyme classifier, ECPred, uses representatives from >50% sequence-similarity clusters to create a non-redundant dataset with only 55 180 enzyme sequences. A consequence of using a smaller dataset is that ECPred is only capable of making 634 complete EC number predictions and 224 partial EC number predictions in contrast with almost 5000 complete EC number predictions in DeepEC.
In addition to being sequence-based, all four ML enzyme classifiers use some variation of a level-by-level approach. Level-by-level predictors decompose the problem into simpler tasks by using one or more predictor for each EC number level. Generally, a binary classifier is used for level zero predictions, which is trained on enzyme sequences for positives and non-enzyme sequences for negatives. From there, level-by-level predictors progress through the EC number hierarchy. The success of level-by-level prediction is due to the diversity criteria considered throughout the EC hierarchy. For example, isomerases are further divided into types of isomerization, a criterion that can only be considered at level two for the isomerase enzyme class. A complete level-by-level strategy is computationally intensive. ECPred trains a different model for each of its 859 possible predictions, making it the most computationally expensive of the four tools. mlDEEPre builds on the level-by-level predictor in DEEPre by adding a model between level 0 and level 1 that is trained to predict if an enzyme has one or multiple EC numbers (because it catalyzes multiple reactions). Different reactants are the most frequent reason for assigning multiple EC numbers to one enzyme (Dönertaş et al., 2016); therefore, mlDEEPre can also be useful for substrate identification which we describe below.
There are a number of difficulties in creating direct comparisons among the most recent models. The evolutionary nature of biological data complicates evaluations. Most enzymes within a category are homologs and can differ by only a few amino acids which are irrelevant to the enzymatic activity. Training and then testing on close homologs prevents evaluations from being based on truly new data, thereby producing overfit and inflated metrics because at least part of the test set is similar to the training set. Although methods that check sequence similarity, such as ECPred and DEEPre, report more reliable self-performance evaluations, differences in sequence similarity cutoffs used complicate comparing these evaluations. Studies frequently also include comparisons with competing tools. To improve comparability, studies often evaluate their performance on a test set created by previous methods. When compared on a common test-set, (Roy et al., 2012) ECPred and DEEPre display similar performance.
New, specialized test sets are also useful for performance evaluations, such as the no Pfam set created by the ECPred authors. Since Pfam annotations are added using homology, the no Pfam test-set represents challenging cases closer to those faced when predicting on novel and de novo sequences. ECPred outperforms DEEPre when compared on the set with no Pfam annotations. However, as acknowledged by ECPred’s authors, DEEPre’s performance may be hindered by its use of domain annotations as a feature, which illustrates how such tests cannot directly compare competing algorithms. Considerations for accurate comparisons include differences in how models report classifications. For example, DeepEC only generates and evaluates complete four level comparison. Conversely, ECPred predicts only partial EC numbers in cases where a fuller prediction is at low confidence or is not possible. Thus, for four-level EC numbers that ECPred predicts, ECPred surpasses DEEPre. Whereas for complete four-level prediction that included classes ECPred does not predict, DEEPre shows superior metrics. Finally, EC numbers are frequently added, amended, transferred and deleted based on new data. To our knowledge, no ML tools re-train with every EC number update to account for this. An extreme example of this issue is that most recent publications ignore translocases, a new seventh level one enzyme class added in mid-2018.
ML for catalytic site prediction
Catalytic site prediction may assist in computationally discriminating between better and worse enzyme designs. Unlike enzyme classification which is best suited to sequence information, structure-based methods work best for predicting catalytic sites. Structure-based methods require annotations at the residue level, as opposed to EC number whole protein level annotations. The Mechanism and Catalytic Site Atlas (M-CSA) contains residue level annotations for 964 unique reactions, making it the best available database (Ribeiro et al., 2018). These annotations can only be mapped to active sites for ~ 60% of the 91 112 protein structures in the protein databank with associated EC numbers (as of 11/19/2020) (Berman et al., 2000).
Enzyme site predictor PREvaIL (Song et al., 2018) passes both sequence and structural features to a random forest (RF) algorithm that is capable of ranking features by their importance to the model thereby revealing features contributing to enzyme activity. Top features for PREvaIL came from both sequence—such evolutionary information—and structural information like solvent accessibility and contact number. PREvaIL’s authors compared PREvaIL to other methods using eight different data sets. Competitors included support vector machines (SVMs) and neural networks that use sequence and/or structural data (Gutteridge et al., 2003; Petrova and Wu, 2006; Zhang et al., 2008). For a holdout test set, PREvaIL has recall and precision values of 62.2% and 14.9% respectively, which is better than a sequence based SVM’s recall and precision values of 50.1% and 14.7% respectively (Zhang et al. 2008). When contextualizing these low precision rates, it is important to note the extreme imbalance of data used for catalytic residue prediction. High percentages of non-catalytic residues in the set increase the difficulty of attaining high levels of precision. PREvaIL outperformed competing methods with one exception: a neural network method with similar overall performance. However, the neural network only performed similarly to PREvaIL when the network included both sequence and structure features followed by spatial clustering (Gutteridge et al., 2003).
A new type of feature extraction method for enzyme site prediction was recently implemented by the Altman group (Torng and Altman, 2019). In this method, local atomic distribution, represented by a grid-like data structure is used as input for a convolutional neural network (CNN). CNNs are a deep learning algorithm known for their excellent performance on spatial applications such as facial recognition. Hence, passing the raw structural data allows the CNN to perform feature extraction which can overcome issues faced by physiochemical properties, such as data loss and high dimensionality. The three-dimensional CNN (3DCNN) outperformed a CNN which was passed physicochemical features, calculated by the author’s commonly used FEATURE program (Bagley and Altman, 1995). Unfortunately, this new feature extraction method has only been shown to produce models capable of predicting a specific enzyme domain or family. As such, it will require additional work relative to previously discussed tools and is limited to use for enzyme domains with available data. The 3DCNN has an impressive average recall of 95.5% and a precision of 99%. When comparing these metrics to PreVAIL’s metrics, it is important to note that the 3DCNN models cover a single enzyme class connected to one M-CSA. Conversely, PreVAIL is intended to cover all enzymes, testing and training on proteins with less than 30% sequence similarity making for few, if any, M-CSA reactions being present in both the training and testing sets.
Our lab has recently developed MAHOMES (Feehan et al., 2021), which predicts if a metal binding site is catalytic or non-catalytic. Focusing on metal-binding sites drastically lowered the level of imbalance between enzymatic and non-enzymatic data. Furthermore, similarities in the local environment of all protein bound metal ions enabled MAHOMES to home in on features responsible for catalytic activity. MAHOMES achieved a 92.2% precision and 90.1% recall using exclusively structural-based features describing physiochemical properties, making it an attractive option for selecting candidates in de novo enzyme design. We benchmarked MAHOMES against three sequence-based, enzyme function predictors, including DeepEC (Ryu, Kim and Lee, 2019) and DEEPre (Li et al., 2018). MAHOMES displayed the best overall performance even though the evaluation data was designed to be particularly challenging for MAHOMES, containing no homology to any data used for training. The development of MAHOMES considered 14 binary classification algorithms. Decision-tree ensemble methods (like those used by PreVAIL) were found to be superior. Decision tree ensemble methods also allowed for analysis of feature importance. The top features described electrostatic perturbations and the number of the residues lining the site. These electrostatic perturbation features had previously been shown to be predictive of catalytic residues by Ondrechen and colleagues (Tong et al., 2009; Somarowthu et al., 2011).
Applications of Enzyme Properties Prediction
Predicting enzymatic function and the level of activity for that function are important steps in enzyme engineering and must be taken into account in order to engineer or select enzymes that are useful in industrial settings. Enzyme activity is dependent not only on the specificity of an enzyme for different substrates, but also the enzyme activity dependence on the microenvironment in which the enzyme performs its function. This microenvironment includes many factors, such as the reaction temperature and enzyme solubility. ML has been used to predict optimal enzyme conditions, enzyme substrate specificity and has aided in understanding which properties of enzymes are impacted most by changes in the microenvironment in which enzymes function.
Condition optimization
Each enzyme works best at a specific temperature and most enzymes must be soluble in specific media in order to function. These conditions are critical for large-scale industrial use of biocatalysts. ML methods are an attractive bioinformatics tool for determining optimal enzyme conditions. Current models exclusively use sequence-based features (and not structure-based features), thereby reducing the cost and complexity in dataset creation.
Recent ML models with features of single- or pairs-of-amino acids have shown compelling successes in determining optimal temperatures for organismal growth and catalysis (Li et al., 2019a, b). Using a non-linear regression vector machine model, organismal growth temperature was predicted from sequence data and then subsequently used in a random forest model as a feature, along with amino acid composition to predict the optimal catalytic temperature for individual enzymes (Li et al., 2019b). For this second task, combining amino acid composition features with a physiological property such as optimal growth temperature provided higher prediction performance. Interestingly, attempts to improve the model with further feature engineering and the introduction of a more complex deep learning model (a CNN) (Li et al., 2020) did not produce higher performance in predicting enzyme catalytic temperature. The CNN likely underperformed other methods due to the small size of training data as the model attempts to automatically extract features from the data instead of relying on features obtained from domain knowledge or from other embeddings.
Significant improvements to the original organismal growth temperature model (Li et al., 2019b) were obtained when data set imbalance was reduced (Gado et al., 2020). This work was informed by the fact that available data on temperature-dependent enzyme function is highly skewed, with most experimental data points normally distributed around 37°C, and less than 5% of data having temperatures above 85°C. This skew had led to overall low performance when predicting high temperature enzymes despite the considerable use of enzymes at high temperatures in industry. The model was improved using imbalance-aware ML models in combination with resampling and ensemble learning. The best performance of the resampling model was obtained with an ensemble tree-based model, which had a 60% improvement in prediction of optimal catalytic temperature at high temperature ranges.
Other features derived from protein sequences such sequence-order information (Chou, 2001) and frequency of amino acid triads (Shen et al., 2007) have also been used for enzyme optimum temperature determination. These features were used to successfully classify a small set of xylanases into three thermophilicity classes (non-thermophilic, thermophilic and hyper-thermophilic) (Foroozandeh Shahraki et al., 2020). Thus, the state of the art allows for accurate prediction of optimal temperature conditions (both growth and catalytic) from sequence data alone, at least within classes of enzymes.
Determining optimal conditions for industrial enzyme use also includes enzyme solubility as experimental characterization usually requires proteins to be soluble in heterologous expression. EnzymeMiner is a pipeline that includes a ML model for solubility prediction (Hon et al., 2020). The model predicts solubility of an enzyme in an Escherichia coli expression system and the whole pipeline is available as a webserver. Using a template-based approach, EnzymeMiner produces a ranked list of candidates for experimentation from a list of putative-enzymes of unknown function. The EnzymeMiner pipeline identifies enzymes from user-defined criteria and ranks the results according to the predicted solubility. EnzymeMiner can be used as a downstream step after models that predict catalytic temperature to prioritize sequences for experimental studies. As a webserver, EnzymeMiner allows for interoperability of ML models. Since several isolated tools (described above) predict optimal conditions for different aspects of the microenvironment where enzymes function, selection guides such as EnzymeMiner that promote easy-to-run integrated workflow should receive more focus in future ML efforts for enzyme engineering.
Substrate identification
Enzymes can be promiscuous—they can catalyze changes on multiple substrates with varying specificities. Substrate identification for enzymatic activities is a subset of the larger field of drug and small molecule identification (Lo et al., 2018). When coupled with high-throughput experimental technologies and deep evolutionary analysis, ML can help identify enzyme substrates with particular specificities or affinities for a variety of enzymes.
Peptide substrates have been designed to bind to specific 4′-phosphopantetheinyl transferases while not binding to homologs using a method called POOL (Tallorin et al., 2018). In this design, an interpretable Naive Bayes classifier allowed for iterative improvement and was therefore preferred to black-box methods like SVMs or deep neural networks.
ML has also been used to identify which glycosyltransferases attach which sugars (Taujale et al., 2020). Evolutionary deep mining identified regions of sequences responsible for the sugar transfer mechanism and transferase acceptor specificity. While the latter is determined by hypervariable regions in the enzyme sequence and are subfamily specific, the former is determined by features of the binding pocket. Expanding on previous work with a smaller glycosyltransferase family (Yang et al., 2018), an ensemble tree method was trained to identify and annotate donor substrate for a large portion of glycosyltransferases with unknown function. Interestingly, as in the case of temperature optimization, ensemble learning provided high prediction performance in a hard task with extensive sequence variation. This six-substrate classification problem achieved 90% accuracy predicting the transfer of six different sugars. Similar to the model in Tallorin et al. (2018), the ensemble Gradient Boost Regression Tree model allowed for interpretability by measuring relative importance of different features. Specific conserved residues (including second shell residues) were identified as determinants for specificity. Also, physicochemical properties of side-chain volume, polarity and residue accessible area were correlated with each different donor class.
Substrate enantioselectivity has been improved using Innov’SAR (Cadet et al., 2018). Innov’SAR uses the Fourier transform of sequence-based features, which are input into various regression models to predict and improve protein property changes. The Innov’SAR method (which has also been applied in the optimization of other enzyme properties as described below) was able to reduce the size of mutant libraries required for successful directed evolution of epoxide hydrolase enzyme stereoselectivity. Similar enantioselective enrichment was also achieved using model-free optimization (Li et al., 2019a, b). This indicates the possibilities for ML or model-free methods in aiding the process of directed evolution to tune enzymes for desired enantiomers.
The prediction of substrate specificity and identification of protein features that correlate with it have also been applied to less well-characterized enzyme families such as the thiolase superfamily (Robinson et al., 2020a) and adenylate-forming enzymes (Robinson et al., 2020b). Random forest models outperformed other approaches (e.g. artificial neural networks, linear models, SVMs) on these tasks, where enzymes can present extensive sequence variation and have a broad range of substrate specificities. In both cases, amino acids lining the active site were selected and encoded with physicochemical properties. For thiolases, features derived from the substrate and from the whole sequence were also used, and the binary classifier predicted whether an enzyme–substrate pair would be active or inactive (Robinson et al., 2020a). For the adenylate-forming family, evolutionary analysis was combined with ML to detect substrate binding residues and the physicochemical properties of those residues were encoded as features (Robinson et al., 2020b). Substrate identification is closely related to function prediction for multifunctional enzymes, and the methods reviewed in this section complement the deep learning model mlDEEPre described previously (Zou et al., 2019).
Turnover rate
In addition to predicting if a particular thiolase can convert a particular substrate, the level of activity for each sequence/substrate combination was predicted using a random forest regression model (Robinson et al., 2020a). This is an example of a third important application of ML in enzyme engineering: enzyme activity level prediction. This method can be used to compare the relative importance of specific residue locations that are altered in different sequences. This study trained two random forest models: a classifier to decide whether a sequence/substrate pair has enzymatic activity, and a regression model to predict level of activity of a sequence/substrate pair predicted to be active. The most important features for the classifier model included chemical descriptors of the substrate such as aromaticity index and molecular connectivity index. For the activity level prediction, there was a correlation of activity level with oxygen content and molecular connectivity index of substrate. Important protein features included the physicochemical properties of residues lining the pocket.
For the enzyme glucose oxidase (used in food industry and blood glucose measuring devices) ML was successfully applied using the Innov’SAR approach to predict effects of multiple mutations on enzyme activity under different environmental conditions, such as pH and mediator molecules (Ostafe et al., 2020). Using the Innov’SAR approach the model captured the effect of multiple mutations. In addition, the Innov’SAR approach has recently been used to study stability of limonene epoxide hydrolase to identify combinations of mutations that produce proteins less prone to aggregation (Li et al., 2021). The idea behind the Innov’SAR approach of creating a protein spectrum from sequence allows this ML method to determine effects of mutations holistically including the effects of multiple simultaneous mutations on protein properties. Other examples of ML applications to directed evolution of enzymes have recently been reviewed (Yang et al., 2019).
ML models based on molecular dynamics trajectories are also predictive of enzyme reactivity. Features computed from the conformational trajectory of an enzyme-substrate complex were used to predict reactivity of ketol-acid reductoisomerase. Using the least absolute shrinkage and selection operator (LASSO) feature selection (Tibshirani, 1996), features of the reactants at the beginning of the trajectory were shown to be highly predictive of reactivity. Predictive features included intra-substrate features (molecule organization such as bond lengths and angles) and features measuring the interaction between the substrate and the environment (binding and distance to water molecules, metal ions) (Bonk et al., 2019). A method for computing atomistic features similar to the previously described 3DCNN for enzyme site prediction (Torng and Altman, 2019) was trained using molecular dynamics trajectories and was applied to predict reaction rates from hydrolysis trajectories (Chew et al., 2020). Though the enzyme substrate-complex was most predictive of enzyme rates, features quantifying the interaction of the substrate with the environment were also used and shown to partially encode the necessary information for reliable rate prediction. Intra-substrate features were not used in this model leaving open the possibility that previously implemented intra-substrate features (Bonk et al., 2019), could account for the reported missed predictions in this model.
Systems engineering
In addition to optimizing or selecting particular enzymes, ML has been deployed in the improvement of biosynthetic pathways. By changing expression rates, turnover rates and post translational modification rates within metabolic systems, ML can significantly increase cellular production of particular metabolites. These current models point the way to a future that includes the optimization of complete living systems.
The combination of ensemble ML methods with mechanistic genome-scale metabolic models was used to optimize the biosynthetic pathway of tryptophan metabolism (Zhang et al., 2020). Optimizing for combinations of six promoter genes controlling five upstream enzymes of the shimitake pathway improved tryptophan production 74% over already optimized strains. The training dataset used, similar to the previously discussed POOL method (Tallorin et al., 2018), was obtained with high-throughput techniques. Further, these two methods represent a tendency in balancing ML exploitation—a greedy approach where the best option is always taken—with exploration—where suboptimal paths are first explored in search for better final outcomes. The exploit/explore trade-off in ML is easy to see in reinforcement learning, where a machine learning ‘agent’ declines immediate rewards to search for possibly higher gains later in the task. But as exemplified by these two methods, this trade-off can also be applied in a supervised learning setting. Here it was achieved by incorporating a measure of prediction noise in the prediction task, augmenting the space of reachable solutions compared to the exploitative approach. When explicitly comparing exploitation and exploration, the exploitative approach selected promoter combinations that yielded higher metabolite production, while the exploratory approach recommended more diverse promoter combinations improving designs with a subsequent optimization round (Zhang et al., 2020).
Identifying the relative concentrations of upstream enzymes that lead to higher yields and rates in biosynthetic pathways is particularly important for cell-free systems. Artificial neural networks predicted the metabolic flux in the upper part of glycolysis using the relative experimentally determined concentrations of four upstream enzymes as input (Ajjolli Nagaraja et al., 2020). While the tryptophan metabolism model (Zhang et al., 2020) did not extrapolate outside the training data, this glycolysis model is designed to specifically perform extrapolation of predictions using artificial data generated by an auxiliary tree-based classifier. This resampling strategy allowed the model to predict enzyme concentrations that yielded flux values up to 63% higher than the ones present in the training data. An ensemble of 10 neural networks was used to screen and rank a large combinatorial space for the 6-step biosynthetic pathway of n-butanol in Clostridium (Karim et al., 2020). The ML recommendations improved production of two related metabolites by 6- and 20-fold over previously engineered designs with the highest reported yields.
Understanding metabolism through mechanistic genome-scale models is difficult due to lack of experimental data on enzyme turnover numbers, which are costly to obtain and prone to noise. Enzyme turnover numbers using ML were predicted for whole metabolic pathways and improved downstream metabolic inferences on a genome-scale metabolic model. Predictions were based on a combination of features (structure, network, biochemistry and assay conditions) and correlated well with data both from in vivo and in vitro experiments (Heckmann et al., 2018).
As an alternative to more common supervised and unsupervised learning methodologies, reinforcement learning, was applied to study the problem of post-translational regulation of biosynthetic enzymes (Britton et al., 2020). This ML model was used to contradict a common understanding about biological regulation, with the ML model indicating that regulation pushes reactions to a non-equilibrium state with the goal of maintaining the solvent capacity of the cell. Use of ML in this manner demonstrated that such models can be used for biological understanding in addition to engineering improvements.
Enzyme Engineering
Recent work has begun to examine the previously unexplored potential for using machine-learning to design enzymes, either by creating new orthologous enzymes or by designing enzyme function in otherwise non-functional structural scaffolds. There have been two successes in this area, one using sequence features and one using structural features. Direct coupling analysis of sequence data was used to engineer orthologs of chorismate mutases (Russ et al., 2020). A generative ML model was developed based on evolutionary data to successfully sample the sequence space of orthologs of chorismate mutase, a small and essential bacterial enzyme. The small size of the enzyme kept the sequence space small and the essential function of the enzyme allowed for screening using a functional assay. Relative conservation was a key feature used in the generation to preserve critically functional residues. Data generated by the functional assay were then incorporated into a classifier on top of the original generative model. Some sampled generated-ortholog sequences reproduced the activity seen with native chorismate mutases, showing comparable catalytic power. They found 30% of the new designs had sufficient chorismate mutase activity to confer survival, with successful designs showing between 42 and 92% sequence identity to their closest chorismate mutase homolog. This result demonstrates that sequence-based features such as amino acid composition and residue correlations carry sufficient information for design of small enzyme orthologs when a functional screen is available.
In a second design, structural features were used to design aldehyde deformylating oxigenase activity into non-enzymatic helix bundles of the ferritin family (Mak et al., 2020). Using a logistic regression model, a set of structural features was ranked to identify the minimal set of structural features required to recreate this function in non-functional scaffolds. The regression was interpretable and the most important features were energy of the active site, overall system energy and active site volume.
Outlook and Challenges
In enzyme engineering, design and selection, the best feature type and ML method depends on the problem being tackled (Fig. 1, Table I). Sequences are the most simplistic and available protein representation and as such continue to be useful for creating features for every problem discussed here. However, purely sequence-based predictors have only shown to be sufficient for enzyme classification. The problem of enzyme classification is centered around EC numbers, one of the oldest, continuously updated bioinformatics repositories. The abundance of data in that resource has allowed for enzyme classification to transition to deep learning ML methods.
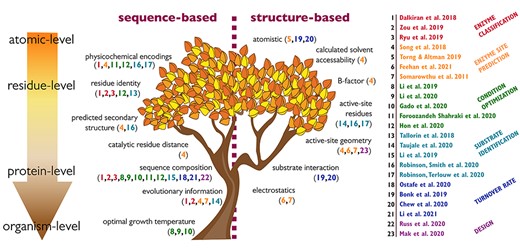
The feature tree. ML models reviewed in this paper according to the type of features used. The maroon dotted line across the center of the tree divides features that can be broadly classified as sequence-based features (left of line) and structure-based features (right of line). The downward arrow categorizes features hierarchically, from atomic (top) to organismal (bottom). Citations for the models (numbered column on the right) are color-coded by the relevant task they are used for (enzyme classification (red), enzyme site prediction (orange), condition optimization (green), substrate identification (teal), turnover rate (blue) and design (purple).
We expect that deep learning methodology will continue to advance the field. Attention learning recently produced a landmark achievement for using sequence data for structure prediction (AlphaFold2.0 unpublished). We believe that the level-by-level architecture of the EC classification problem is particularly well suited to attention learning. Additionally, as more data become available, we are likely to see protein sequences encoded using deep learning embeddings of the type used in natural language processing to produce highly informative feature sets for downstream protein tasks (Alley et al., 2019; Heinzinger et al., 2019). By using embeddings as inputs, simpler models can outperform more complex models relying on data representations, as shown for protein function prediction (Villegas-Morcillo et al., 2020).
All models beyond EC classification, benefit from inclusion of structure-based or experiment-based features. However, data availability for these features remains limited—this is currently being addressed by the community. For example, more than 50 journals currently instruct their authors to use the standards for reporting enzymology data (STRENDA) guidelines (Tipton et al., 2014). As the STRENDA database grows, its inclusion of pH, temperature, Km and Kcat will be crucial for advancing condition optimization, substrate identification and turnover rate predictors.
Another strategy for dealing with limited data is using classical ML algorithms instead of deep learning. Currently, the most commonly used, classical ML algorithm is the tree-based random forest algorithm, which is most suitable for complex classification tasks with high dimensional data including organismal growth temperature prediction, optimal enzyme temperature prediction, enzyme site prediction, substrate classification and activity level prediction.
Ensemble models and the reduction of imbalance in datasets play key roles in model performance as shown with enzyme temperature optimization. Combining predictors showed success over individual predictors, indicating that a better balance between exploration (combination of models) and exploitation (selecting best predictions) is useful to achieve higher performance in enzyme system engineering. It is likely that the future will include more enzyme design efforts being guided by ML, combining ML predictors in more complex workflows in order to reduce screening costs and numbers of rounds of testing in experimentation.
With respect to data representation, no consensus yet exists about the best encoding. A protein function is dependent on its three-dimensional structure and functional sites are known to be regulated by local interactions, solvent accessibility and conformational flexibility (Mazmanian et al., 2020). Deriving these physicochemical properties from the linear amino acid sequence alone can be accomplished by homology modeling followed by calculation of the physicochemical properties (Robinson et al., 2020a). However, a more common strategy is to use the sequence to generate evolutionary information, where conserved sequence positions can enable identification of crucial residue interactions without any structural knowledge. ML models will continue to benefit from continued use of both physicochemical and conservation encodings. An exciting emerging paradigm for enzymatic mechanisms identifies the importance of protein dynamics and networks of residue interactions (Agarwal, 2019) that could guide future encodings.
Finally, despite the difficulties in comparing ML models, even those that perform the same task, such as enzymatic classification, more comprehensive comparisons that specifically evaluate the benefits of each type of feature (evolutionary conservation, structural data, sequence encodings) and feature encoding (one-hot, physicochemical encoding, unsupervised embeddings) will be welcome, since their impact on different protein prediction tasks is an under analyzed question.
Acknowledgements
We gratefully acknowledge helpful discussions with Meghan W. Franklin as well as funding from National Institute of General Medical Sciences award DP2GM128201.
References
Martín Abadi, Ashish Agarwal, Paul Barham. et al. (
Author notes
Ryan Feehan and Daniel Montezano contributed equally.

{kind=link}
{kind=link}