

Abstract
A novel class of intracellular chloride channels, the p64 family, has been found on several types of vesicles. These channels, acting in concert with the electrogenic proton pump, regulate the pH of the vesicle interior, which is critical for vesicular function. Here we describe the molecular cloning of p64H1, a p64 homolog, from both human and cow. Northern blot analysis showed that p64H1 is expressed abundantly in brain and retina, whereas the other members of this family (e.g., p64 and NCC27) are expressed only at low levels in these tissues. Immunohistochemical analysis of p64H1 in rat brain, using an affinity-purified antibody, revealed a high level of expression in the limbic system—the hippocampal formation, the amygdala, the hypothalamus, and the septum. Immunoelectron microscopic analysis of p64H1 in hippocampal neurons demonstrated a striking association between p64H1 and large dense-core vesicles (LDCVs) and microtubules. In contrast, very low p64H1 labeling was found in perikarya or associated with small synaptic vesicles (SSVs) in axonal profiles. Immunoblot analysis confirmed that p64H1 is colocalized with heavy membrane fractions containing LDCVs rather than the fractions containing SSVs. These results suggest that p64H1-mediated Cl− permeability may be involved in the maintenance of low internal pH in LDCVs and in the maturation of LDCVs and the biogenesis of functional neuropeptides.
- p64
- p64H1
- NCC27
- intracellular chloride channel
- large dense-core vesicle
- neuropeptide
- hippocampus
- microtubules
Intracellular chloride channels have been found on several types of vacuolar organelles, including brain clathrin-coated vesicles (Xie and Stone, 1988; Mulberg et al., 1991), kidney endocytotic vesicles (Bae and Verkman, 1990), thyroid parafollicular cell secretory granules (Barasch et al., 1988), and Golgi membranes (Glickman et al., 1983). Chloride conductance may balance the entry of protons generated by the vacuolar H+-ATPase and thus facilitate intravesicular acidification. The resulting acidic vesicle interior is critical for vesicle loading, enzymatic processing of vesicle contents, and membrane trafficking (Bradbury and Bridges, 1994).
Two types of secretory vesicles coexist in nerve endings: large dense-core vesicles (LDCVs; 70–100 nm in diameter) and small synaptic vesicles (SSVs; ∼40 nm in diameter). LDCVs, counterparts of endocrine secretory vesicles, are assembled at the soma and transported via axons to nerve endings. Neuropeptides and some biogenic amines are sorted into LDCVs and released extrasynaptically. In contrast, classical neurotransmitters are released at synapses from SSVs. LDCVs and SSVs have different regulatory mechanisms for neurotransmitter exocytosis (De Camilli and Jahn, 1990). After their release of vesicle contents, LDCVs move by retrograde transport to the soma for reloading (Thureson-Klein and Klein, 1990); SSVs are recycled by endocytosis at the terminal (Winkler, 1997). Both types of vesicles contain proton ATPases (Rudnick, 1986). Because the coexistence of proton pumps and chloride conductance is a universal finding in vacuolar compartments (Al-Awqati, 1995), chloride channels also may be expressed on neuronal secretory vesicles. However, the identities of the vacuolar chloride channels on these vesicles are unknown.
The first cloned intracellular chloride channel, p64, originally was purified from bovine kidney microsomes (Landry et al., 1993). Antibodies to p64 can eliminate chloride conductance on these microsomes (Redhead et al., 1992). Outwardly rectifying anion channel activity was recorded in a reconstituted planar lipid bilayer fused with cell membranes from p64 transfected cells (Edwards et al., 1998). Three p64 homologs—human nuclear chloride channel (NCC27; Valenzuela et al., 1997; Tulk and Edwards, 1998), rat p64 homologue 1 (p64H1;Duncan et al., 1997), and ch loride intracellular channel 2 (CLIC2; Rogner et al., 1996)—have been cloned recently. The sequences of these homologs predict 25–30 kDa proteins that resemble the C-terminal half of p64. For NCC27 and p64H1 the outwardly rectifying anion channel activity was recorded on planar lipid bilayers fused with membranes from transfected cells (Valenzuela et al., 1997; Duncan et al., 1997). CLIC2 activity has not yet been reported. The in vivo distribution and function of the p64-like channels remain unknown.
In the present report we describe the cloning of human and bovine p64H1. To seek the function of p64H1, we examined its cellular and subcellular distributions in those brain regions in which it is highly expressed. Our findings suggest that p64H1 in brain is associated with LDCVs, but not SSVs. We propose that p64H1 may regulate intragranular pH in LDCVs, which is essential for the enzymatic processing and release of functional peptides from LDCVs.
MATERIALS AND METHODS
Isolation of p64H1 gene and plasmid constructs.Partial sequences of bovine p64H1 initially were isolated from the two-hybrid screening of a bovine retinal cDNA library by using pDB-Rho39Tr, which contains the C-terminal 39 residues of human rhodopsin as bait (Chuang and Sung, 1998). For antibody production and purification, constructs encoding glutathione S-transferase (GST) and maltose binding protein (MBP)–p64H1 fusion proteins were generated by inserting the BamHI/XhoI fragment encompassing the coding sequence of the 158 C-terminal residues of bovine p64H1 to the 3′-end of the open reading frames of GST and MBP inBamHI/XhoI-digested pGE5X-2 vector (Amersham Pharmacia Biotech, Arlington Heights, IL) andBamHI/SalI-digested pMAL-cRI* vector, respectively. pMAL-cRI* (Chuang et al., 1995) is a modified pMAL-cRI vector (New England Biolabs, Beverly, MA).
Full-length human p64H1 cDNA was obtained by screening a human retinal λgt10 library, using a 32P-radiolabeled fragment of the bovine sequence as a probe. Library screening was performed at moderate stringency with standard methods. Full-length human p64H1 cDNA was PCR-amplified from purified phage clones, using PCR with primers flanking the insert (forward, 5′-CGCGGATCCAGCAAGTTCAGCCTGGTTAAGT; reverse, 5′- CCGCTCGAGCTTATGAGTATTTCTTCCAGGGT). Then this PCR fragment was subcloned into pGEX-5X-2, pBluescript II KS (pBS II KS; Stratagene, La Jolla, CA), and pCIS (Genentech, South San Francisco, CA) for sequencing, T7 promoter-driven expression, and cytomegalovirus (CMV) promoter-driven expression, respectively. Multiple clones were sequenced to verify that no PCR errors were introduced.
Full-length human NCC27 cDNA (accession number AF109197) also was isolated during the human retinal library screening, using bovine p64H1 cDNA as a probe. Sequencing confirmed that the sequence of NCC27 that we isolated is identical to that reported by others (Valenzuela et al., 1997). The DNA fragment containing the entire coding region of NCC27 was inserted into pMAL-cRI vector and pBS II KS vector for MBP fusion protein production and in vitro translation expression, respectively.
RT-PCR was used to obtain the p64 coding sequence (accession numberAF109199). Bovine retinal poly(A+) RNA was first reverse-transcribed (Superscript II kit, Life Technologies, Gaithersburg, MD) and then PCR-amplified with a pair of primers (forward, 5′-CG GGATCCAGACCTACTCGGGTCTGCTTGC; reverse, 5′-CG GAATTCAGGATCGGCTGAGGCGCTTG) that flank the coding sequence of bovine p64. PCR products were digested withBamHI/EcoRI and then inserted intoBamHI/EcoRI-digested pGEX-5X-2 for both sequence confirmation and GST fusion protein production. Full-length p64 cDNA also was inserted into pBS II KS for in vitrotranslation.
Production and affinity purification of the p64H1 antibodies.All GST (Amersham Pharmacia Biotech) and MBP (New England Biolabs) fusion proteins were produced and purified according to the manufacturer’s instructions. Purified GST–p64H1 fusion protein was used as an immunogen for the production of rabbit antiserum (Cocalico, Reamstown, PA). To remove cross-reactivities, we passed the immunized serum sequentially through three CNBr-activated Sepharose CL-4B columns conjugated with Escherichia coli DH5α lysate, GST protein, and MBP protein. The final flow-through was affinity-purified on a MBP–p64H1 Sepharose column, eluted with 0.1m glycine, pH 2.8, and neutralized with Tris-Cl, pH 9.5. The resulting affinity-purified antiserum is referred to as CUMC29. To remove any further cross-reactivity against p64 and NCC27, we passed CUMC29 over two Sepharose columns, the first conjugated with GST–p64 and the second with MBP–NCC27. The flow-through is referred to as p64H1 antibody in this report (see Results for antibody specificity).
In vitro translation and immunoprecipitation. Bovine p64, human NCC27, and human p64H1 were transcribed and translated individually in reticulocyte lysates in the presence of35S-methionine (NEN, Boston, MA) according to the manufacturer’s instructions (TNT T7 Quick coupled transcription/translation system, Promega, Madison, WI). The translated products were examined first by SDS-PAGE analysis. Equivalent amounts of 35S-labeled p64H1, NCC27, and p64 translated products were mixed, diluted in 1% Triton X-100 and 0.5% bovine serum albumin (BSA) containing STE buffer (50 mm Tris, pH 7.5, 150 mm NaCl, and 2 mm EDTA), and immunoprecipitated by protein A-Sepharose conjugated with either CUMC29 or p64H1 antibody for 4 hr at 4°C. Then the beads were washed with Triton X-100 containing STE buffer once, STE buffer without Triton X-100 once, and eluted with Laemmli sample buffer before SDS-PAGE analysis and autography.
Northern blotting. Twenty micrograms of total RNA isolated from various bovine tissues (RNAzol, TEL-TEST, Friendswood, TX) were electrophoresed on 1% formaldehyde agarose gel, transferred, cross-linked to nylon membrane, and incubated in prehybridization buffer (5× SSC, 30% formamide, 2% SDS, 200 μg/ml denatured herring sperm DNA, and 5× Denhart’s) at 42°C for 2 hr before the [α-32P]-dCTP-labeled (NEN) bovine p64H1 probe (containing the C-terminal 158 residues and the entire 3′-untranslated region) was added for an additional overnight incubation at 42°C. The blot was washed three times at room temperature with 1× SSC and 0.1% SDS before autoradiography. After exposure, the blot was stripped (0.05× SSC, 10 mm EDTA, and 0.1% SDS at 80°C for 30 min twice, followed by 0.01× SSC for 15 min at room temperature) and reprobed, first with 32P-labeled full-length p64 cDNA and subsequently with NCC27 cDNA.
Light microscopic immunocytochemistry. All immunocytochemical methods were approved by the Weill Medical College of Cornell University Institutional Care and Use Committee. Six Sprague Dawley rats (Taconic, Germantown, NY) were used in the light microscopy immunocytochemical experiments. All rats were anesthetized with Nembutal (150 mg/kg, i.p.) and sequentially perfused through the ascending aorta with (1) normal saline (0.9%) containing 1000 U/ml of heparin (10–15 ml); (2) 50 ml of 3.75% acrolein (Polysciences, Warrington, PA) and 2% paraformaldehyde (PFA) in 0.1 mphosphate buffer (PB), pH 7.4; and (3) 200 ml of 2% PFA in PB. The brains were removed, and coronal blocks (5 mm thick) were cut and stored in 2% PFA for an additional 30 min. Sections (40 μm thick) were cut on a vibratome, collected in PB, and then placed in 1% sodium borohydride in PB for 30 min to remove excess aldehydes (Veznedaroglu and Milner, 1992). Immunocytochemistry was performed as described previously (Hsu et al., 1981). In brief, the sections were incubated with p64H1 antibody (1:6000 in the presence of 0.25% Triton X-100 and 0.1% BSA) overnight at room temperature and then for 24 hr at 4°C. Sections were rinsed and incubated with biotinylated goat anti-rabbit IgG for 30 min, followed by peroxidase–avidin complex (Vector Laboratories, Burlingame, CA) for 30 min before the diaminobenzidine substrate color development. To demonstrate the specificity of the antibody, we preadsorbed the immunoreactivity by incubating diluted primary antibody (1: 2000) with 10 μg of MBP–p64H1 fusion protein overnight at 4°C. The supernatant, collected after centrifuging the sample at 12,000 rpm for 15 min, was used for immunostaining as described above. The stained sections were mounted on acid-cleaned slides previously coated with 1% gelatin. They were air-dried, dehydrated, and coverslipped with DPX (Aldrich Chemical, Milwaukee, WI).
Electron microscopic immunocytochemistry. For electron microscopy, sections through the hippocampal formation of two rats were prepared for immunoperoxidase labeling as described above, except that 0.035% Triton X-100 was used in the primary antibody diluent. Additionally, sections through the hippocampal formation of three rats were prepared for the immunogold method. For this, the rats were pretreated with a zinc chelator (sodium diethyldithiocarbamate; Fluka, Ronkonkoma, NY) to diminish silver background labeling (Veznedaroglu and Milner, 1992) before perfusion with fixative. The sections were processed by the “freeze–thaw” technique and then incubated in p64H1 antibody (1:250 dilution) for one overnight incubation at room temperature and an additional 24 hr at 4°C. After several washes the sections were incubated with goat anti-rabbit IgG conjugated to 1 nm gold particles (1:50 dilution; AuroProbe One, Amersham Pharmacia Biotech) in 0.1% gelatin and 0.8% BSA in PBS for 2 hr at room temperature. Sections were rinsed with PBS, post-fixed in 1.25% glutaraldehyde in PBS for 10 min, and rinsed once in PBS, followed by 0.2 m sodium citrate buffer, pH 7.4. The conjugated gold particles then were enhanced by treatment with silver solution (IntenSE, Amersham Pharmacia Biotech) for 6–8 min (Chan et al., 1990). The sections were fixed with 2% osmium tetroxide for 1 hr, dehydrated, and embedded in Epon 812. As described previously (Milner and Veznedaroglu, 1992), ultrathin sections (50 nm thick) then were prepared and counterstained before examination on a Philips CM10 electron microscope. Sections containing the hippocampal formation regions obtained from three different rat brains were analyzed electron microscopically. The nomenclatures used in this study are according toPeters et al. (1991).
Isolation and fractionation of synaptosomal vesicles.Synaptosomal vesicles were isolated from rat cerebral cortices (including the hippocampal formation) and fractionated exactly as described (Huttner et al., 1983). Briefly, isolated crude synaptosomes were lysed hypotonically, and the postmitochondrial membranes were pelleted by centrifugation (165,000 × g for 2 hr). The resulting pellet (LP2) was resuspended in a total of 1.7 ml of 40 mm sucrose. The suspension was subjected to 10 up-and-down strokes in a glass Teflon homogenizer spun at 1200 rpm and then passed through a 25-gauge needle five times. The LP2 pellet suspension was layered on top of a 10.3 ml continuous 50–800 mm sucrose gradient and then centrifuged for 5 hr at 65,000 × gin an SW41 rotor (Beckman Instruments, Palo Alto, CA). The gradient, except for the pellet, was collected in 1 ml fractions from the bottom of the tube. The density of each fraction was measured by refractometry. Of each fraction, 0.5 ml was precipitated with 10% (v/v) trichloroacetic acid (14,000 × g for 15 min at 4°C), followed by two washes of ice-cold acetone. The collected pellets were resuspended in 40 μl of Laemmli buffer. Aliquots were loaded on 4–20% gradient SDS-PAGE (Novex, San Diego, CA) for immunoblotting assays. Blots were probed with p64H1 antibody (1:500), calcium-dependent activator protein for secretion (CAPS) antibody (1:500; Berwin et al., 1998), or anti-synaptophysin p38 monoclonal antibody (1:2000; Jahn et al., 1985).
Immunoblotting. Human embryonic kidney 293s cells were transfected by using calcium phosphate methods as previously described (Sung et al., 1991). 293s cells transfected for 1 d were homogenized and lysed in STE buffer containing 1% Triton X-100 and protease inhibitor cocktail (1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 0.7 μg/ml pepstatin). The supernatant was centrifuged at 14,000 × g for 15 min, separated by 4–20% gradient SDS-PAGE, and transferred to nitrocellulose membrane for immunoblotting. Immunodetection was performed with either the Proto-Blot system (Promega) or the chemiluminescent method (Pierce Chemical, Rockford, IL).
RESULTS
Cloning of the bovine and human p64H1
In a yeast two-hybrid screen for proteins interacting with the C terminus of rhodopsin, several positive clones were isolated from a bovine retinal library. A GenBank search using BLAST revealed that some of these positive clones contained sequences similar to members of the p64 family. Although the interactions between these isolates and the C terminus of rhodopsin were specific, the physiological relevance of these interactions is not the scope of this study and will be presented elsewhere.
To obtain a full-length cDNA sequence of this p64-like protein, we screened a human retinal cDNA library. The longest cDNA clones that were isolated contained an open reading frame encoding a protein of 253 amino acids with a predicted molecular weight of 28.6 kDa and a predicted pI of 5.3 (Fig.1A; accession numberAF109196). The reading frame was flanked by 78 bp of 5′-untranslated sequence and a 1796 bp 3′-untranslated region. The sequence around the first ATG conformed with the Kozak sequence for translation initiation sites (Kozak, 1991). The deduced amino acid sequence of these human cDNAs shared highest overall identity (∼95%) to rat p64H1 (accession number AF104119), which also encodes for a 253 residue peptide. Our cDNA clones also shared high similarity to human NCC27 (64% identity; accession numbers U93205 and AF109197), human CLIC2 (62% identity; accession O15247; data not shown), and the C-terminal half of bovine p64 (72% identity; accession numbers L16547 and AF109199) (Fig.1B). Although the bovine cDNA (accession numberAF109198) initially isolated from the two-hybrid screening was not full-length, its deduced amino acid sequence was 94 and 95% identical to the corresponding regions of human and rat p64H1, respectively, and was much less similar to NCC27 and p64 (Fig. 1B). The high degree of conservation between our human and bovine clones and rat p64H1 unambiguously identified our clones as the bovine and human orthologs of rat p64H1.

Sequence and alignment of the human p64H1.A, Nucleotide sequence of human p64H1 cDNA clone, including the full deduced amino acid coding sequence. The putative transmembrane domains are underlined. The potential tyrosine phosphorylation site is circled.B, Alignment of partial bovine p64H1 amino acid sequence with full-length human (H) p64H1, rat (R) p64H1, human NCC27, and bovine (B) p64, using the Jotun–Hein algorithm. Identical amino acids are indicated in the boxed regions. Note that the N-terminal sequences of bovine p64 were omitted for the sequence alignment.
Hydropathy analysis suggested that p64H1, like p64, may have one or two putative transmembrane domains (Fig. 1A, underlining). The existence of the second transmembrane helix is questionable; although its hydrophobicity and length are characteristic of a transmembrane domain, the presence of several charged residues in this region may disqualify it. Further analysis will be required to confirm the topology of p64H1. In agreement with Duncan et al. (1997), the four potential protein kinase C and one protein kinase A consensus sites found in rat p64H1 also were found in human p64H1. However, we noted an additional consensus sequence for tyrosine phosphorylation: tyrosine 244 was flanked by an acidic residue at its −3 position and an alkaline residue at its −6 position (Hunter, 1982; Patschinsky et al., 1982). Interestingly, although this putative tyrosine phosphorylation site was conserved evolutionarily among rat, bovine, and human p64H1, it was not present in either p64 or NCC27. Similar to rat p64H1, no obvious signal peptide sequence or N-glycosylation site was found in human p64H1.
Generation and specificity of the p64H1 antibody
A GST fusion protein containing the C-terminal 158 residues of bovine p64H1 was used to immunize rabbits for the production of antibody against p64H1. The immunized serum was affinity-purified against MBP fused with the antigenic epitope of p64H1, and the resulting serum was referred to as CUMC29. CUMC29 recognizes bacterially expressed recombinant GST–p64H1 fusion, but not GST alone (data not shown). To remove potential cross-reactivity, we passed CUMC29 sequentially through Sepharose columns conjugated with GST–p64 and MBP–NCC27 fusion proteins. The flow-through activity will be referred to as p64H1 antibody. We predict that CLIC2 cross-reactivity, if any, should be removed by this immunodepletion also, because 96% of the identical amino acid residues shared by p64H1 and CLIC2 in the epitope region are also present in both p64 and NCC27. Furthermore, the nonidentical residues of CLIC2 and p64H1 in this region share less similarity than the other two proteins.
The p64H1 antibody recognized a single band with an apparent molecular weight of ∼29 kDa in rat brain (Fig.2A, lane 1) on immunoblots. This protein band comigrated with the recombinant human p64H1 protein overexpressed in 293s cells (lane 2) as well as with in vitro-translated p64H1 (lane 3). The identical molecular masses of endogenous and recombinant p64H1 indicated that our isolated cDNA clones contained the entire open reading frame. Although the predicted molecular masses of NCC27 and p64H1 are very similar, in vitro-translated NCC27 migrated slightly but detectably more slowly than p64H1 on the same SDS-PAGE (lane 4). This was consistent with a previous observation that NCC27 had an aberrantly slower mobility on SDS-PAGE (Tulk and Edwards, 1998). Thus, the signal recognized by p64H1 antibody was clearly not NCC27.

Specificity of the p64H1 antibody.A, Protein extracts from rat brain (lane 1) and 293s cells expressing p64H1 (lane 2) and also in vitro-translated proteins p64H1 (lane 3), NCC27 (lane 4), and p64 (lane 5) were electrophoresed on a single SDS-PAGE and transferred to nitrocellulose. One-half of the membrane (lanes 1–2) was cut and immunoblotted with the p64H1 antibody; the remaining membrane (lanes 3–5) was visualized by autoradiography.B, Similar amounts of radiolabeled in vitro-translated p64H1, NCC27, and p64 were mixed, and a small aliquot was removed for direct SDS-PAGE analysis (lane 1). The remainder was divided into two fractions and immunoprecipitated by either protein A-Sepharose conjugated with CUMC29 (lane 2) or p64H1 antibody (lane 3). The immunocomplex was eluted, separated on SDS-PAGE, and visualized by autoradiography.
To be certain that the p64H1 antibody was specific to p64H1 and did not cross-react with NCC27 and p64, we subjected a mixture of similar amounts of 35S-labeled in vitro-translated P64, NCC27, and p64H1 (Fig. 2B, lane 1) to immunoprecipitation by either p64H1 antibody or CUMC29. The p64H1 antibody precipitated p64H1, but not p64 or NCC27 (Fig.2B, lane 3). In contrast, the original affinity-purified CUMC29 precipitated p64H1, p64 to a lesser extent, and very little NCC27 (Fig. 2B, lane 2). Therefore, the p64H1 antibody was used in all subsequent experiments.
p64H1 is expressed abundantly in the nervous system
A Northern blot was performed to determine the tissue distribution of p64H1. The probe was a 32P-labeled DNA fragment encoding the C-terminal coding region and the 3′-untranslated region of bovine p64H1. A single ∼5 kb transcript was detected in total RNA isolated from many bovine tissues. High levels of p64H1 messenger were found in heart, lung, retina, and brain (Fig.3, top panel). Equivalent amounts of RNA were loaded as judged by the ethidium bromide-stained agarose gel before blotting (Fig. 3, inset). The same RNA blot then was stripped and reprobed with full-length clones of bovine p64 (Fig. 3, middle panel) and human NCC27 (Fig. 3, bottom panel). A ∼6 kb transcript of p64 was expressed at high levels in heart and lung, and a ∼1.7 kb transcript of NCC27 was expressed at high levels in kidney, lung, and spleen. The probing of CLIC2 was omitted, because a previous report showed that CLIC2 was expressed at low levels in some tissues such as fetal liver and adult skeletal muscle; these signals are only detectable by RT-PCR, but not by Northern blot assay (Rogner et al., 1996). The unique transcript size of each of these p64 homologs was in agreement with that reported previously. Minor variability is seen in the relative expression levels among different tissues. However, p64 and NCC27 consistently were found to be expressed at relatively low levels in the brain, where p64H1 is in high abundance. The enriched expression of p64H1 in both brain and retina indicated that p64H1 may be involved functionally in the CNS.

Northern blot analysis of p64H, NCC27, and p64. Total RNA isolated from the indicated different bovine tissues was electrophoresed and transferred. The same RNA blot was probed sequentially with radiolabeled cDNA fragments of bovine p64H1 (top panel), bovine p64 (middle panel), and human NCC27 (bottom panel). The RNA molecular weight standards are indicated (in kb; left). Inset, Photograph of ethidium bromide-stained agarose gel.
The cellular and subcellular localizations of p64H1 in the rat brain
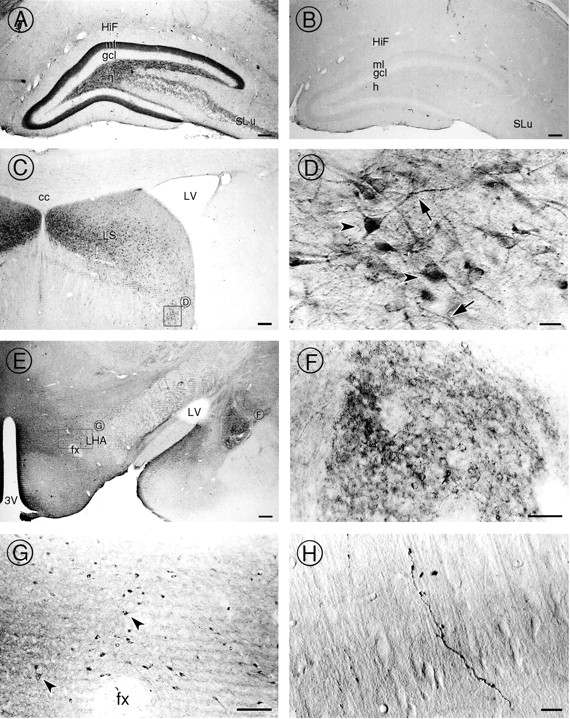
To gain insight into the functions of p64H1, we examined the cellular and subcellular distribution of p64H1 immunoreactivity in rat brain. Light microscopic examination of the brain revealed intense p64H1-immunoreactive processes in several “limbic” areas, including the hippocampal formation (Fig.4A), the septum (Fig.4C,D), the amygdala (Fig. 4E,F), and the hypothalamus (Fig. 4E,G). In addition, certain areas in the cortex (Fig. 4H) and the cerebellum (data not shown) were also p64H1-positive. In these regions, p64H1 immunoreactivity was associated mainly with fibers that had several different appearances. In the hippocampal formation a dense plexus of p64H1-immunoreactive processes was detected in the inner molecular layer of the dentate gyrus. Large, punctate p64H1-labeled processes were found in the mossy fiber pathway in the hilus of the dentate gyrus and stratum lucidum of the CA3 region (Fig.4A). In the central amygdala the p64H1 immunoreactivity was in fibers that formed baskets outlining the somata (Fig. 4F). On the other hand, p64H1 labeling in the cortex regions was located mainly on single, large varicose fibers (Fig. 4H). In addition to processes, p64H1-labeled somata were observed in the lateral septal nuclei (Fig.4D) and the lateral hypothalamic nuclei (Fig.4G). All labeling was specific and could be ablated efficiently by preincubating the antibody with the fusion protein that was used as antigen; one example taken from the hippocampal formation region is demonstrated (Fig. 4B).

Light microscopic localization of p64H1 in several regions of rat brain. A, In the hippocampal formation, strong immunoperoxidase activity was detected in the inner molecular layer (ml) and hilus (h) of the dentate gyrus and in stratum lucidum (SLu) of the CA3 region. In contrast, the granule cell layer (gcl) lacked any p64H1 labeling.HiF, Hippocampal fissure. B, After preadsorption of the antibody with fusion protein containing the antigenic epitope, no p64H1 labeling was observed in any region. The hippocampal formation region is shown as an example.C, Strong p64H1 labeling was seen in neurons of the lateral septal (LS) nuclei region. cc, Corpus callosum; LV, lateral ventricle.D, High magnification of the lateral septal nuclei region shows intense p64H1 immunoreactivity in the somata (arrowheads) as well as in processes that have a beaded appearance (arrows). E, The hypothalamus and amygdala also contain intense p64H1 immunoreactivity in varicose processes.3V, 3rd ventricle; fx, fornix;LHA, lateral hypothalamic area. F, High magnification of the amygdala shows p64H1 immunoreactivity in baskets of punctate fibers surrounding the somata. G, In the lateral hypothalamic nuclei area, p64H1 also is associated perikarya (arrowheads). H, p64H1 immunoreactivity on single, large varicose fiber was observed in the cerebral cortex. Scale bars: A–C, G, 100 μm; D, 10 μm; E, 200 μm; F, 50 μm;H, 5 μm.
The subcellular localization of p64H1 in the dentate gyrus was examined further by electron microscopy. Consistent with the light microscopic analysis, p64H1 immunoreactivity was confined to axonal profiles in this region. Prominent p64H1 immunoreactivity (as demonstrated by the immunogold-silver reaction product) was associated primarily with LDCVs (∼70–100 nm, Fig. 5A,B). Despite the low abundance of LDCVs in most nerve terminals, the majority of LDCVs in the surveyed fields was labeled with p64H1. Some immunogold-silver particles representing p64H1 immunoreactivity were in close contact with the plasma membrane (Fig. 5A). Immunogold-silver particles also were associated with tubular membranes (Fig. 5B) that may represent the axonal reticulum, a membranous structure often found to be positive for LDCV markers (Quatacker et al., 1992; Annaert et al., 1994). Despite the high abundance of SSVs within synaptic profiles, immunogold-silver particles rarely were found to be affiliated with SSVs (Fig.5A,B).

Electron micrographic localization of p64H1 in the inner molecular layer of the dentate gyrus. A, p64H1 immunoreactivity (as demonstrated by immunogold-silver particles) primarily associated with LDCVs (open arrows). Immunogold-silver particles occasionally were affiliated with the plasma membrane (arrowheads). Very little p64H1 labeling was associated with SSVs in most synaptic profiles. B, p64H1 sometimes was found on tubular membrane profiles (solid arrow). Scale bars, 0.5 μm.
In axons, immunogold-silver particles representing p64H1 labeling often were affiliated with microtubular bundles (Fig.6A,B). In some cases the microtubule-associated p64H1 labeling extended over a distance of ∼8 μm. In some planes of section this labeling was affiliated with LDCVs within axonal profiles (large arrows in Fig.6C).

p64H1 immunoreactivity was associated with microtubules in axons. A, B, A low-magnification photomicrograph showed that p64H1 immunoreactivity (as demonstrated by immunogold-silver particles) was arrayed remarkably along a longitudinal section of microtubule bundles (small arrows). C, A high-magnification photomicrograph revealed that p64H1 was associated with LDCVs (large arrows) that were in direct contact with microtubules. Scale bars, 0.5 μm.
p64H1 is found primarily in higher density membrane fractions
To support our immunohistochemical observation that p64H1 is associated predominantly with LDCVs, we examined the localization of p64H1 in vesicles derived from brain synaptosomes by immunoblotting analysis. As shown in Figure 7, p64H1 antibody recognized a single band of 29 kDa in fractions of high density (1.07–1.09 gm/ml), with a peak in 0.6–0.7 msucrose fractions that was consistent with a previously reported density of LDCVs isolated by a similar method (Coyle and Kuhar, 1974). A LDCV marker, CAPS (Berwin et al., 1998), showed a largely overlapped colocalization with p64H1 in these fractions. In contrast, the SSV marker synaptophysin was highly enriched with a peak in the 0.2–0.3m sucrose fractions (with density from 1.02 to 1.04 gm/ml). This result is in agreement with previous reports of SSV density (Huttner et al., 1983; Jahn et al., 1985).

Distribution of p64H1, the LDCV marker CAPS, and the SSV marker synaptophysin in synaptosomal vesicle fractions. Fractionation was performed on a continuous sucrose density gradient (see Materials and Methods). An aliquot of each fraction was TCA-precipitated and subjected to SDS-PAGE (a 200 μl equivalent of each fraction for p64H1, a 20 μl equivalent for CAPS, and a 1 μl equivalent for synaptophysin). Then the protein gels were transferred and probed with p64H1 antibody, CAPS antibody, or anti-synaptophysin p38 antibody. The sucrose concentration is indicated. Note that CAPS, which has been dissociated from LDCV, was distributed in the loading zone of the gradient.
DISCUSSION
Intracellular chloride channels are heterogeneous
Chloride channels exhibit marked diversity in conductance, current–voltage relationship, and regulation. Three members of the p64-like intracellular chloride channel family (i.e., p64, p64H1, and NCC27) have their own unique transcript size and tissue expression pattern (Landry et al., 1993; Duncan et al., 1997; Tulk and Edwards, 1998; this study). Northern blot analysis revealed that p64H1 is expressed abundantly in both the retina and the brain, where p64 and NCC27 have relatively low levels of expression. On the other hand, some tissues express more than one p64-like protein. For example, p64H1, p64, and NCC27 are highly expressed in the lung. The distinct tissue distributions of the p64 homologs suggest that the expression of each of these molecules is regulated specifically and might be tailored for their respective functional roles in different cell types.
Vacuolar H+-ATPase is composed of several subunits. Although the same H+-ATPase gene products are expressed universally on many vacuolar compartments, the measured proton pump activity is variable in different organelles (for review, see Al-Awqati, 1986; Finbow and Harrison, 1997). It is conceivable that the various vacuolar chloride channels differentially modulate proton pump activity and therefore the degree of acidification. This hypothesis is in agreement with the finding that different organelles show markedly different Cl− requirements for acidification and their subsequent physiological functions (Rudnick, 1986). Ion transport is a highly regulated cellular function. One probable mechanism is via the phosphorylation of channel proteins. Putative protein kinase C, protein kinase A, and tyrosine phosphorylation consensus sites of p64H1 are conserved across species.In vitro studies have demonstrated that translated rat p64H1 is phosphorylated by protein kinase C (Duncan et al., 1997). Nevertheless, whether or not phosphorylation regulates the expression and/or physiological behavior of p64H1 in vivo remains to be determined.
Selective distribution of p64H1 in rat brain
Immunoreactivity for p64H1 was evident in many regions that comprise the “limbic” system (i.e., hippocampal formation, amygdala, and lateral septum). In these regions, p64H1 labeling primarily was confined to fibers and varicose processes. Our electron microscopic analysis of the hippocampal formation demonstrated that most of these labeled varicosities were axons and axon terminals. However, prominent perikaryal labeling of p64H1 was evident in the lateral septal and lateral hypothalamic regions. Previously, p64H1 had been localized to endoplasmic reticulum-like membranes surrounding the nucleus in transiently transfected fibroblasts (Duncan et al., 1997). Whether the p64H1-positive structure observed in these fibroblasts is similar to the perikaryal labeling observed in the present study remains to be examined.
The proposed function of p64H1 in maintaining the low internal pH of LDCVs and neuropeptide biogenesis (see below) is supported by its high level of expression in the hypothalamus, a region in which peptidergic neurons are in high abundance. Moreover, the cellular and subcellular expression pattern of p64H1 is strikingly similar to that of calcitonin gene-related peptide in rats treated with chemical-injected insult (Bulloch et al., 1996). This correlated expression pattern suggests that p64H1 could be involved in the biosynthesis of this or a related peptide.
p64H1 in enriched in LDCVs
Immunoelectron microscopic analysis of the hippocampal formation and the immunoblotting of fractionated synaptosomal vesicles clearly revealed that p64H1 was associated predominantly with LDCVs. These results suggest that p64H1 is involved in functions associated with LDCVs. LDCVs are formed at exit sites of the trans-Golgi network (TGN) as immature secretory vesicles and transported to the release sites, during which maturation takes place. At the Golgi apparatus LDCVs are packaged with neuropeptide precursors, processing enzymes, and members of the secretogranin family (for review, see Burgess and Kelly, 1987;Tooze and Burke, 1987). Low pH and high calcium concentration in the TGN promote protein condensation that selectively excludes other proteins not destined for LDCVs as one mechanism of sorting (Gerdes et al., 1989). After leaving the Golgi apparatus, the electron dense-core continues to mature by progressive aggregation of the vesicle contents, a process that also is promoted by low pH (Tooze and Stinchcombe, 1992). More importantly, to produce functionally active peptide, the neuropeptide precursor has to be cleaved by endo/exoproteases that generally have acidic pH optima (for review, see Gainer et al., 1985). The function of LDCVs is consistent with the notion that the internal pH of LDCVs is acidic, pH 5–6 (Loh et al., 1984). The finding that p64H1 is enriched in LDCVs may suggest that p64H1-mediated chloride permeability, in action with the proton pump, is involved in the generation and maintenance of an acidified microenvironment in LDCVs.
Several enzymes (e.g., acetyltransferase and α-amidating enzymes), which further modify neuropeptides after proteolytic cleavage, show maximal activity at neutral pH and are inhibited at acidic pH (for review, see Gainer et al., 1985). Aggregated proteins also are thought to be solubilized at more neutral pH before release (Tooze and Stinchcombe, 1992; Dartsch et al., 1998). We speculate that the expression or the channel activity of p64H1 may be regulated, probably via phosphorylation of the protein or by interactions with cofactor(s). These changes subsequently affect the intravesicular pH and, in turn, the activities of neuropeptide processing enzymes.
At the electron microscopic level, p64H1 labeling is seen occasionally in close apposition to axonal plasmalemma as well as LDCVs. Although there is the possibility that p64H1 is expressed on the plasmalemma, p64H1 more likely is expressed on LDCVs in the process of exocytosis. Previous experiments that used digitonin-permeabilized nerve endings isolated from neurohypophyses concluded that chloride ions are a crucial component for a late step in exocytosis and that this step can be inhibited by a chloride channel antagonist (Dayanithi and Nordmann, 1989). Further studies are need to determine whether p64H1 has a role in exocytosis.
LDCVs are concentrated in the axonal periphery via microtubule-based anterograde transport along the axon (Tooze and Burke, 1987). However, unlike SSVs, which recycle at the nerve endings, LDCVs also travel back to the cell body for vesicle reloading once their contents are released (Thureson-Klein and Klein, 1990). The microtubule-mediated translocation of LDCVs in axons is consistent with our observation that p64H1 immunoreactivity is associated with microtubules. Both the anterograde and retrograde transport of LDCVs may account for the high-density labeling of p64H1 associated with the microtubules in axonal profiles. Whether p64H1 is involved in this membrane trafficking needs to be determined.
Perspectives
Complementary approaches often are needed to determine the specific function of a novel putative channel protein in vivo. Similarly, although chloride conductance can be recorded on reconstituted lipid bilayers fused with p64H1-expressing membranes, channel activity observed in artificial bilayers does not necessarily reflect a true function in vivo. The p64H1 protein may be a subunit of a chloride channel complex instead of a channel itself, or it may be a channel regulator that activates a channel already present in the membranes used for recordings. Alternatively, p64H1 may behave as both a channel and a regulator. The molecular cloning of p64H1 certainly will help us to better define its functional roles. Mutations found in several chloride channels/channel regulators/transporters are linked to human disease (Simon et al., 1996, 1997; Thakker, 1997). For example, mutations in CLCNKB, a kidney-specific chloride channel, cause type III Bartter’s syndrome that is characterized by renal failure (Simon et al., 1997). Thus, the isolation of human p64H1 will allow us to examine its relationship with human pathological conditions in future studies.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant EY11307, The Foundation Fighting Blindness, and a Cornell Scholarship to C.-H.S.; and by NIH Grants MH42834, DA08259, and HL18974 to T.A.M. We are indebted for the kind gifts of human retinal cDNA library from Dr. J. Nathans (Johns Hopkins University School of Medicine, Baltimore, MD), of anti-CAPS antibody from Dr. T. F. Martin (University of Wisconsin), and of anti-synaptophysin p38 monoclonal antibody from Dr. P. Greengard (Rockefeller University, NY). We thank Drs. Gustavo Frindt and Lawrence Palmer for helpful discussion and A. W. Tai, J. Macke, and Drs. Tim McGraw and J. Wagner for critically reading this manuscript.
Correspondence should be addressed to Dr. Ching-Hwa Sung, The Margaret M. Dyson Vision Research Institute, Weill Medical College of Cornell University, 1300 York Avenue, New York, NY 10021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}